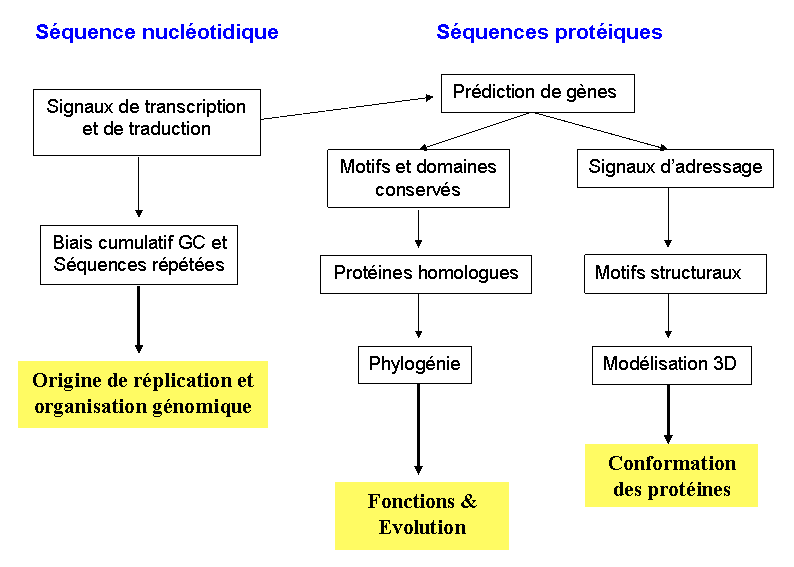
La Bioinformatique
Définition
On regroupe sous le terme
de bioinformatique un champ de recherche multi-disciplinaire
où travaillent de concert biologistes, informaticiens,
mathématiciens et physiciens, dans le but de
résoudre un problème scientifique posé
par la biologie.
La bioinformatique est basée sur des concepts
nécessaires à l'interprétation de
l'information génétique (séquences),
structurale (repliement 3D) et fonctionnelle (activité).
C'est le décryptage de la « bio-information
». La bioinformatique est donc une branche
théorique de la biologie.
Les analyses bioinformatiques permettent d'énoncer des
hypothèses (ex. : comment les protéines se
replient ou comment les espèces évoluent) et de
formuler des prédictions (ex. : localiser ou fonction d'un
gène).
Un 'bioinformaticien' n'est pas le simple croisement d'un biologiste et
d'un informaticien (pas plus qu'un neurochirurgien n'est celui d'un
psychiatre et d'un anatomiste). Ces TP viseront à
découvrir et maitriser certains outils bioinformatiques
couramment utilisés par le biologiste
moléculaire. Adapter l'outil en fonction de la question et
voir les limites de certains logiciels.
Le suffixe 'Informatique' doit donc être compris comme
renvoyant à l'interprétation de 'l'information'
biologique, et non pas à l'utilisation de l'ordinateur.
L'ordinateur n'est qu'un outil permettant de faire des
prédictions fonctionnelles et structurales.
Toutes ces prédictions sont basées sur deux
fondements.
Importance du travail de paillasse
Les prédictions
sont
basées sur les résultats acquis l'invention de la
biochimie et la découverte de l'ADN et de sa structure (il
n'y a
"que" 50ans).
Malgré la qualité des outils disponibles, toute
analyse
bioinformatique ne donne qu'une prédiction in silico,
nécessitant une confirmation in vivo. La biologie
à de
nombreuses fois démontré qu'elle
tolère toutes les
exceptions.
Cette dernière décennie, un lourd investissement
a entrainé une
augmentation des capacités de production
de séquences (voir chapitre
Bases de données) mais la quantité de
données
expérimentales n'arrive pas à suivre cette
vertigineuse
ascension.
Evolution et Homologie
Toutes les prédictions bioinformatiques sont basées sur la recherche et l'existence de parentés entre biomolécules. Deux protéines possédant un ancêtre commun sont dites homologues, et possèdent et caractéristiques communes.
Le postulat de base, basé sur la théorie de l'évolution, veut que des protéines apparentées (dites homologues) possèdent des similarités au niveau de la séquence ADN, de la séquence protéique, de la forme de la protéine et donc fonction biologique.
L'homologie est une propriété qualitative entre des protéines, elle n'est pas quantifiable. Des protéines ne sont pas très, moyennement ou faiblement homologue.
Horloge moléculaire
Des
recherches ont permis de constater que le taux d'accumulation des
mutations dans le génome d'organismes différents
est du
même ordre de grandeur dans des régions homologues
(régions soumises à la même pression de
sélection).
L'accumulation sera maximale pour des régions qui ne sont
pas
soumises à la pression de sélection naturelle (ne
codant
pas pour des gènes) et minimale dans les parties du
génome soumises à une forte pression (c'est
à dire
les régions codant pour des fonctions essentielles
à la
survie de l'organisme).
Chaque séquence accumule les mutations à un
rythme qui
lui est propre et qui est dicté par l'intensité
de la
pression de sélection à laquelle elle est
soumise. Pour
reconstituer des phylogénies (dater la divergence entre deux
espèces), on peut utiliser différentes
molécules
comme on utilise les aiguilles d'une montre pour calibrer l'horloge :
- la trotteuse des secondes (taux de mutation important, par exemple un
pseudogène) pour des évènements
récents
(études des sous populations au sein d'une
espèce) ;
- l'aiguille des minutes (taux de mutation moyen, par exemple le
cytochrome C) pour l'analyse d'un passé proche ;
- l'aiguille des heures (taux de mutations faible : les histones) pour
l'étude d'un passé lointain.
La vitesse d'évolution de la séquence est du
même
ordre de grandeur au sein d'une même classe fonctionnelle de
protéines et elle est différente pour des
protéines qui ont des fonctions différentes : la
vitesse
d'évolution de la sérum albumine est toujours
plus
importante que celle du cytochrome C. Ces différences de
vitesse
dépendent à la fois de la probabilité
qu'une
substitution apparaisse et de sa compatibilité avec la
survie de
l'organisme.
Si l'on admet cette théorie, et que l'on connaît
le taux
d'accumulation des mutations, il est possible d'estimer le temps de
divergences d'espèces en comparant leur diversité
moléculaire.
Organisation de l'information d'une biomolécule
La structure primaire est la séquence. Elle correspond une succession linéaire.
Structure secondaire (ADN et protéine) :
La structure secondaire décrit le repliement local du squelette carboné. La structure secondaire vient du fait que des repliements énergétiquement interviennent localement au sein de la chaine nucléotidique ou peptidique.
- ADN
Les séquences répétées inversées peuvent provoquer des conformations de type tête d'épingle (hairpin).

Ces structures jouent entre autre un rôle dans la
reconnaissance
par des facteurs de réplication et de transcription.
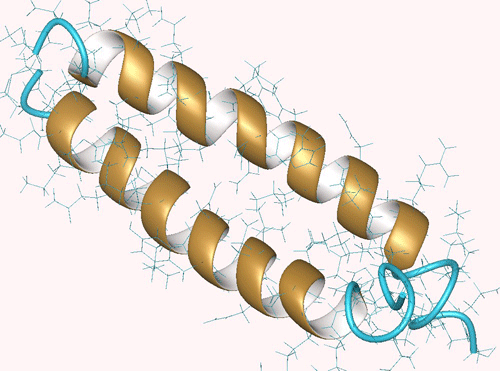
- Protéines
Les hélices a:
Les feuillets B:

 Les coudes
Les coudes
Ils permettent les jonctions entre les hélices a et les feuillets
B.
Structure tertiaire (Protéine seulement) :
C'est le repliement de la chaîne polypeptidique dans l'espace. On parle plus couramment de structure tridimensionnelle, ou structure 3D.
Le concept de base veut une relation entre la structure et la fonction. La structure 3D d'une protéine est intimement liée à sa fonction: lorsque cette structure est cassée par l'emploi d'agent dénaturant, la protéine perd sa fonction: elle est dénaturée. La biologie structurale à montré que certains acide aminés sont beaucoup plus important que d'autre dans la conformation de la protéine. Ces acides aminés clés seront recherchés durant les TP par accès à des banques de motifs, ils sont importants pour la prédiction de la fonction d'un gène et pour déterminer l'importance d'une mutation.
La structure tertiaire d'une protéine dépend de
sa structure primaire.
Ainsi, deux protéines homologues ayant une forte
similarité de
séquence (> 80 % des acides aminés
identiques) auront également des
structures très proches. La prédiction de la
structure tertiaire a
partir de la structure primaire est une des quêtes de Graal
de la recherche actuelle.
La biologie structurale possède certaines limites influençant la structure des protéines déterminées: importance des interactions protéiques, contexte de la protéine (pH, Température, salinité, cofacteur, en présence du substrat, fixé à l'ADN...)
Durant ce TP vous aurez l'occasion de travailler sur des structures de protéines de leur mutants des maladies.
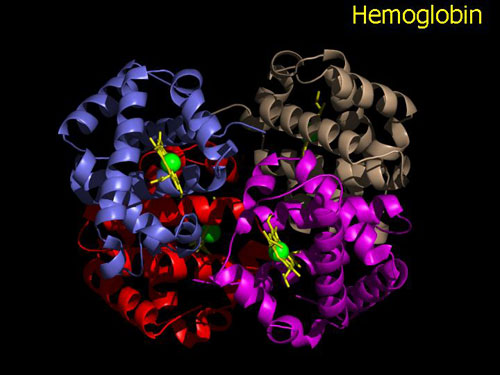
Structure quaternaire (Protéine seulement) :
La structure
quaternaire des protéines regroupe l'association
d'au moins deux chaînes polypeptidiques - identiques ou
différentes -
par des liaisons non-covalentes (liaison H, liaison ionique,
interactions hydrophobes), et parfois des ponts disulfures.
Chacune de ces chaînes est appelée monomère (ou sous-unité) et l'ensemble oligomère ou protéine multimérique.
L'hémoglobine est un exemple de structure quaternaire ; elle est constituée de 4 sous-unités : 2 sous-unités α (de 141 acides aminés) et 2 sous-unités β (de 146 acides aminés).
{kind=link}
Récap
Des recherches ont permis de constater que le taux d'accumulation des mutations dans le génome d'organismes différents